How to dowload stored sequence files from NCBI using KENCLUST
Prepared by Angus A. Nassir
In the last post, I demonstrated How to Transfer Files from your computer to KENCLUST and from KENCLUST to your computer. In this post, I describe how to download sequence files from SRA using SRA Tools. The Sequence Read Archive (SRA) is the largest database of high-throughput sequencing data. This database is maintained by the National Centre of Biotechnology Information (NCBI). Anyone can submit their sequence data to SRA for storage. Types of data stored in SRA include NGS data, metagenomics data and trace archive files. Trace files are DNA sequence chromatograms, base calls, and quality estimates for single-pass reads from various large-scale sequencing projects. Files are stored in a variety of formats including BAM, CRAM, FASTQ, fasta with quality scores, HDF5, SFF among others. See the SRA website for a list of all acceptable formats. These files can be accessed from multiple cloud providers and NCBI servers and used for bioinformatics analysis.
| Step 1: click here to go to the SRA website. |
 |
| Step 2: Enter your search query. |
| For example, we may want to retrieve sequence data for pancreatic cancer. To do this, type the query “pancreatic cancer” on the search field.
|
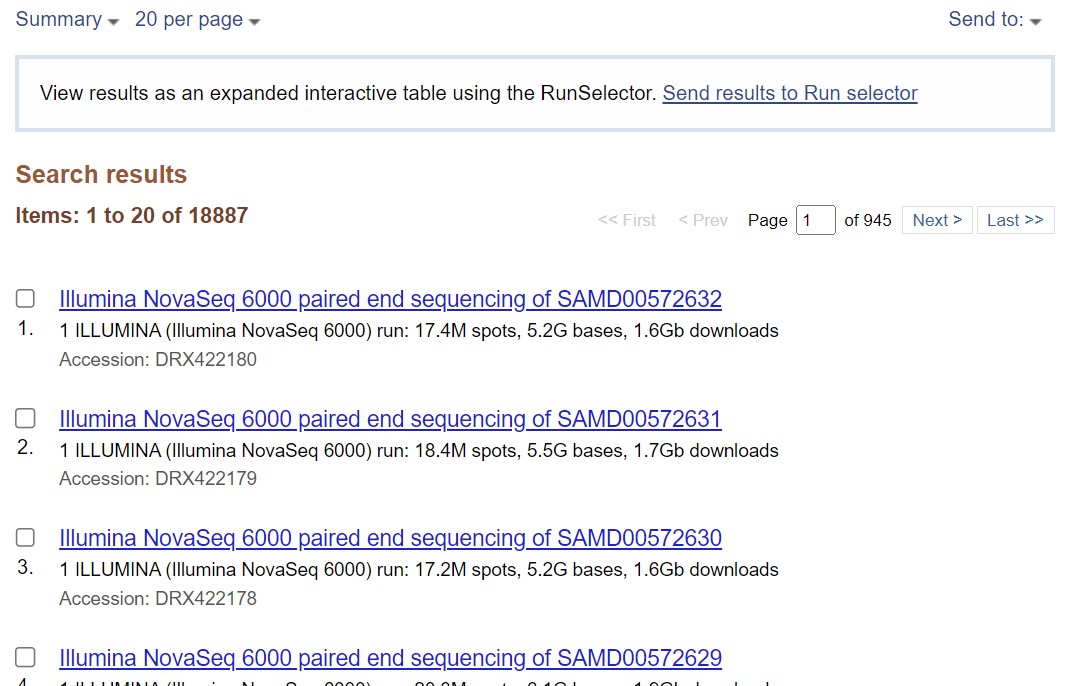
| Sequences associated with the search query will be listed. |
|
|
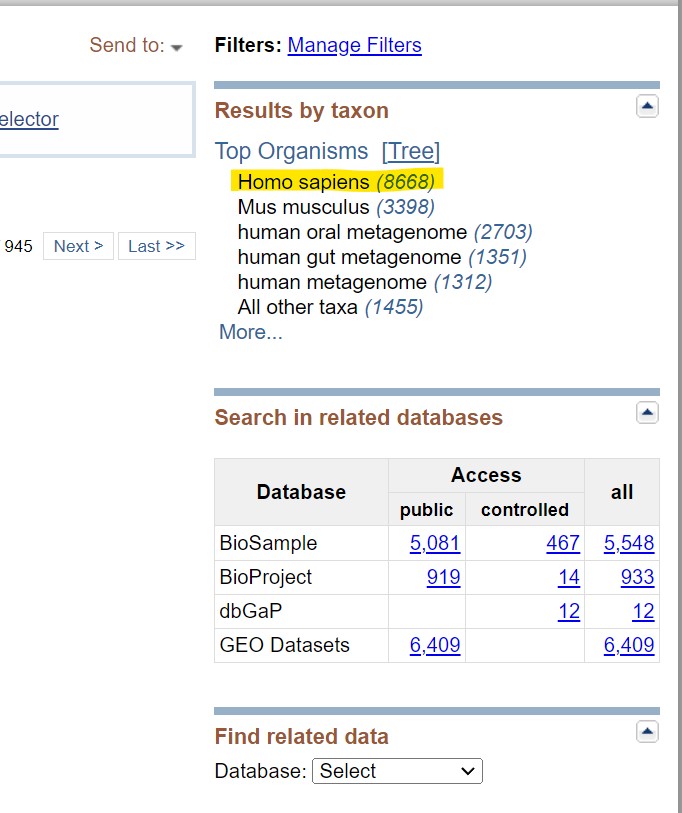
Step 3: Filter results
You can filter these results based on the species type. For example, to get human pancreatic cancer sequences click on the “homo sapiens” link on the top right corner.
 |
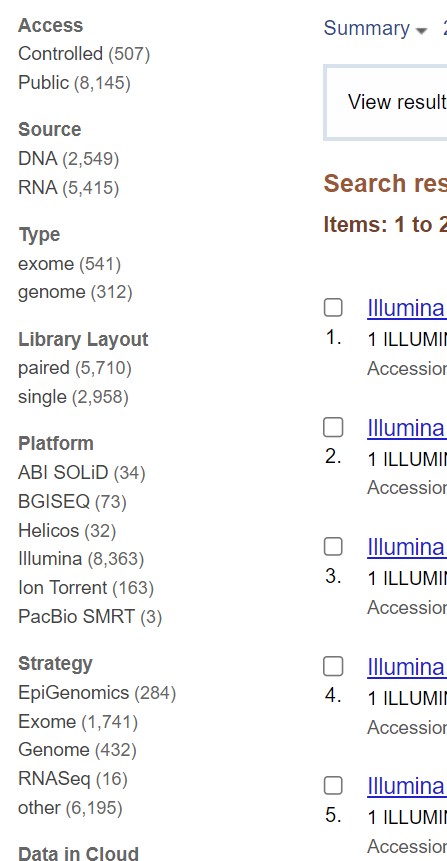
| There are additional filters on the left side of the results page that allow you to further refine your search based on platform used, whether you require DNA or RNA sequences, whether you need single or paired reads, file type, and so on.
|
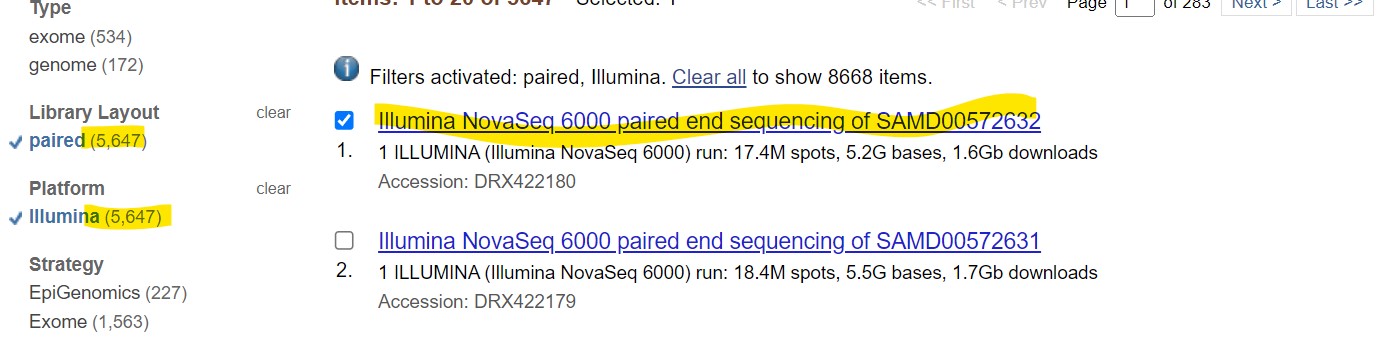
| Step 4: Make your selection |
| Here, we choose illumina paired end reads and select the first result
|
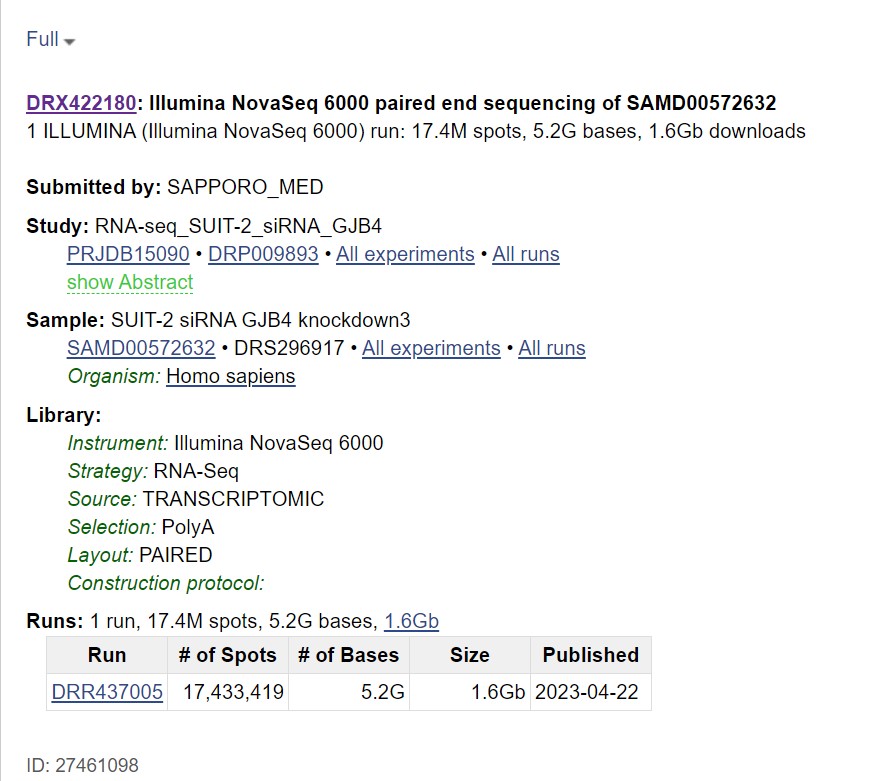
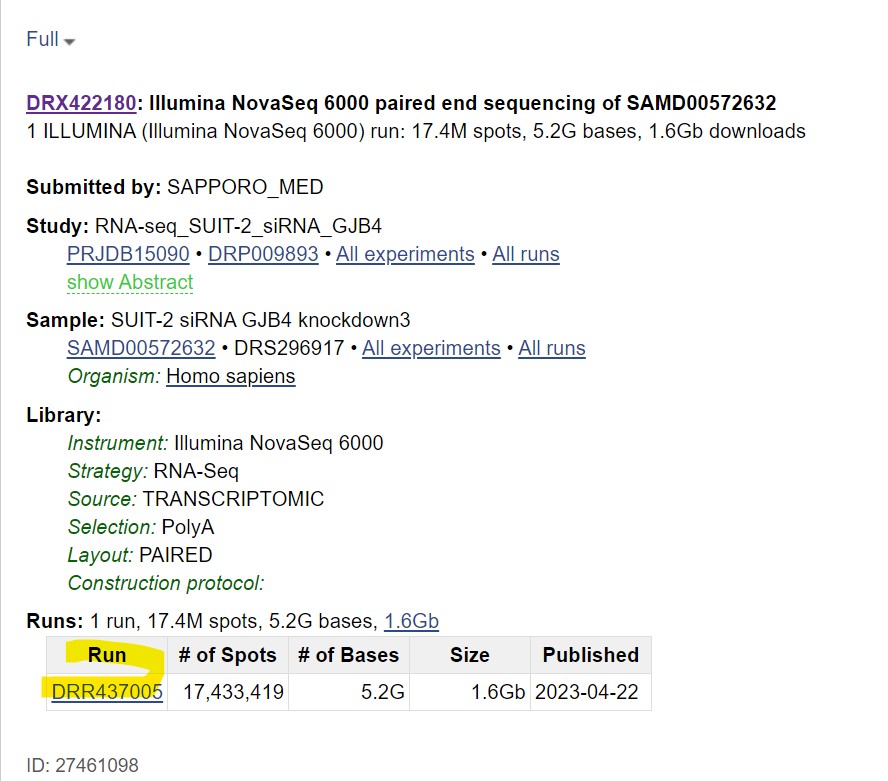
| Step 4: Click on the “run” link at the bottom to access the reads
This gives you the following output:
|
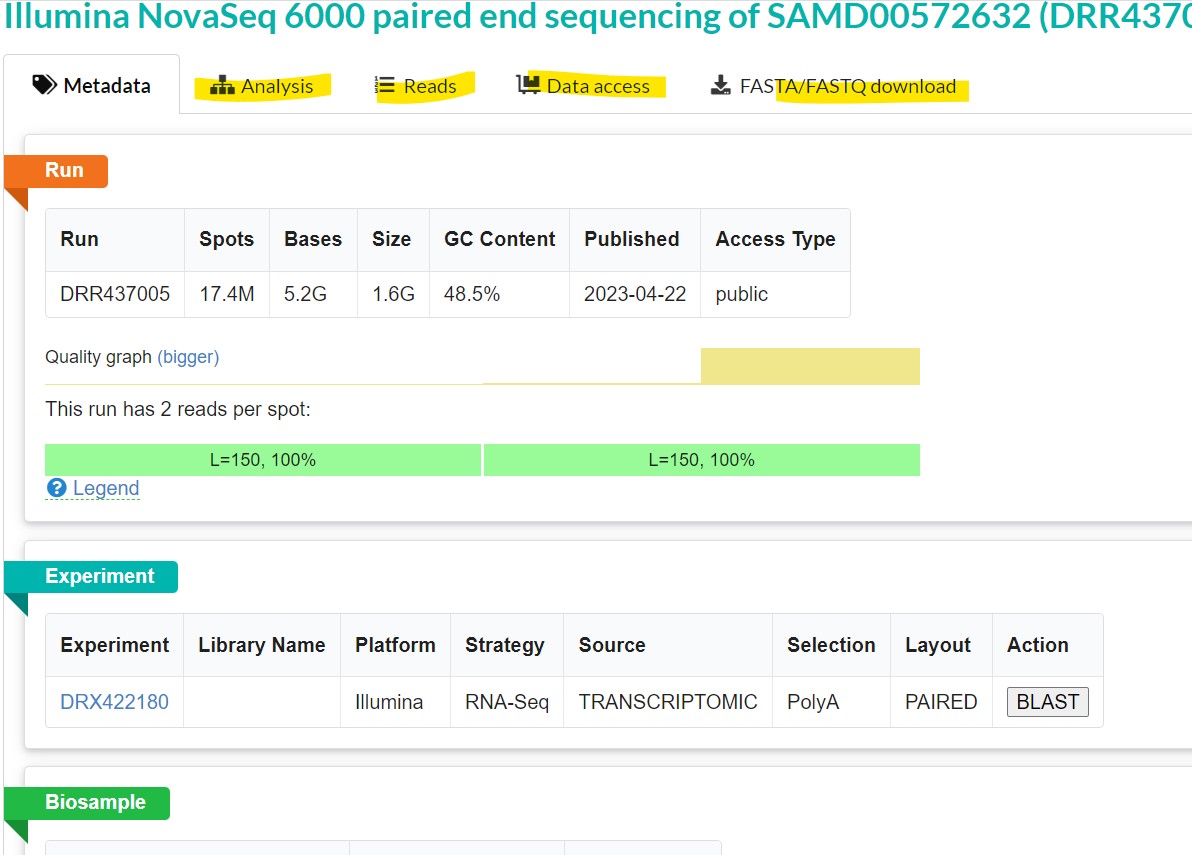
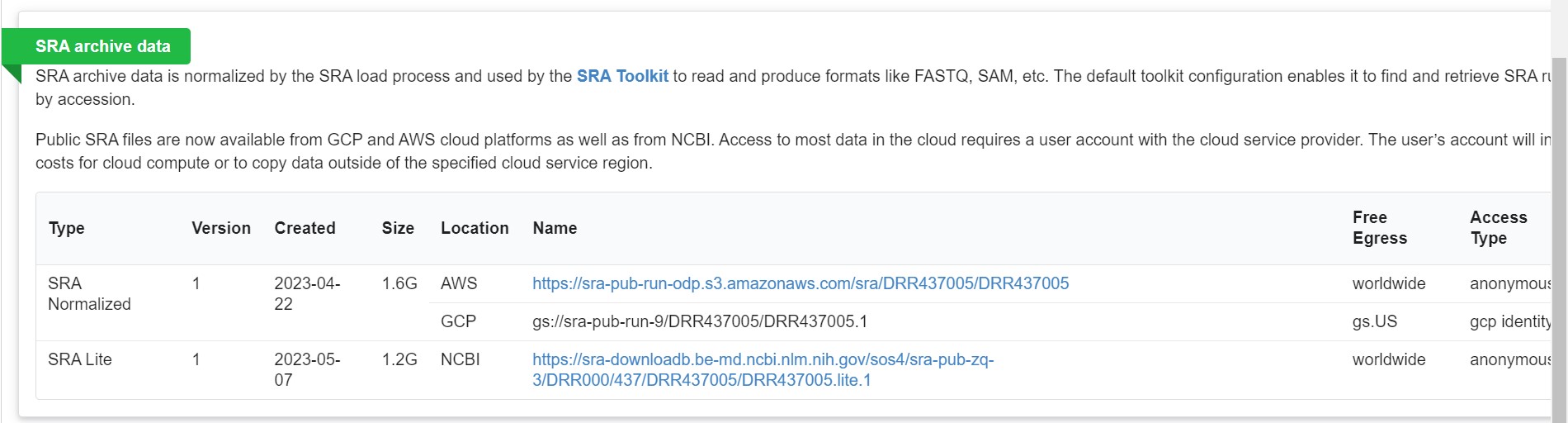
Clicking on the “Analysis” tab gives you taxonomic analysis of the chosen sequences Clicking on “Data access” tab gives you links that allow you to download the data from publicly accessible databases.
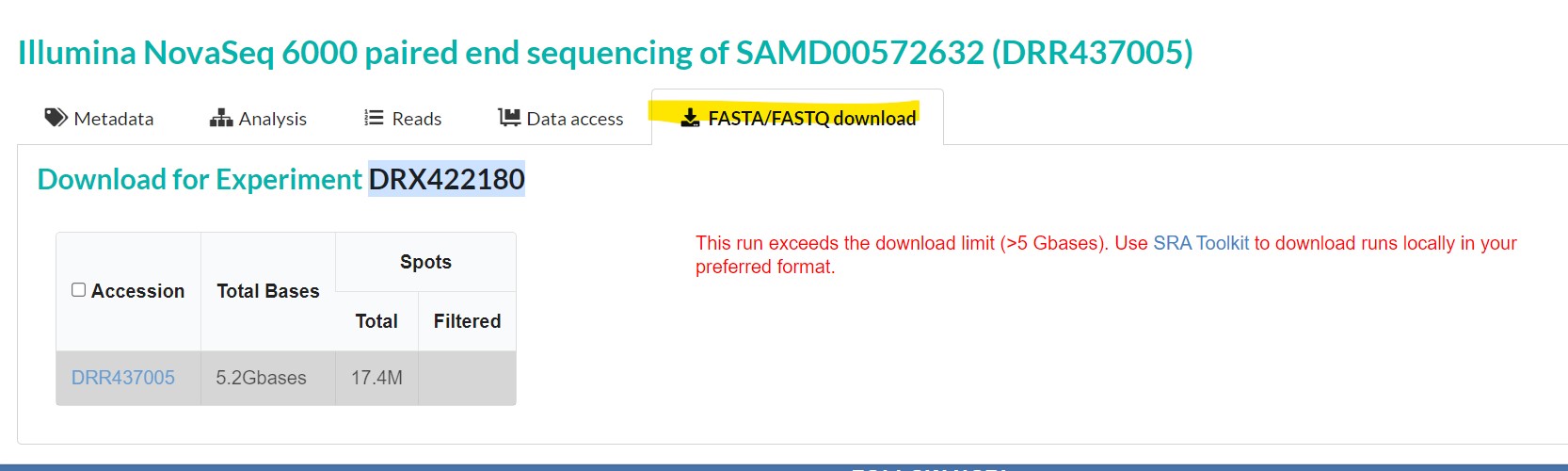
The “fasta.fastq download” tab allows you to download the sequence in your local machine in FASTA or FASTQ format |
| Step 5: Access KENCLUST and use the SRA Toolkit |
|
However, reads exceeding 5GB cannot be downloaded using this method. One has to use the SRA Toolkit to download such reads.
Prefetch DRR437005 |
| Downloads go to the NCBI folder in your root by default and are saved with the .SRA ending. |
| Step 6: Convert SRA file to fastq format
To convert the SRA Convert SRA file to fastq format, use the following command:
fastq-dump DRR437005 This will generate a file called DRR437005.fastq You can rename this file and use it to perform your analysis |
| Step 7: To download single reads instead of the entire sequence file |
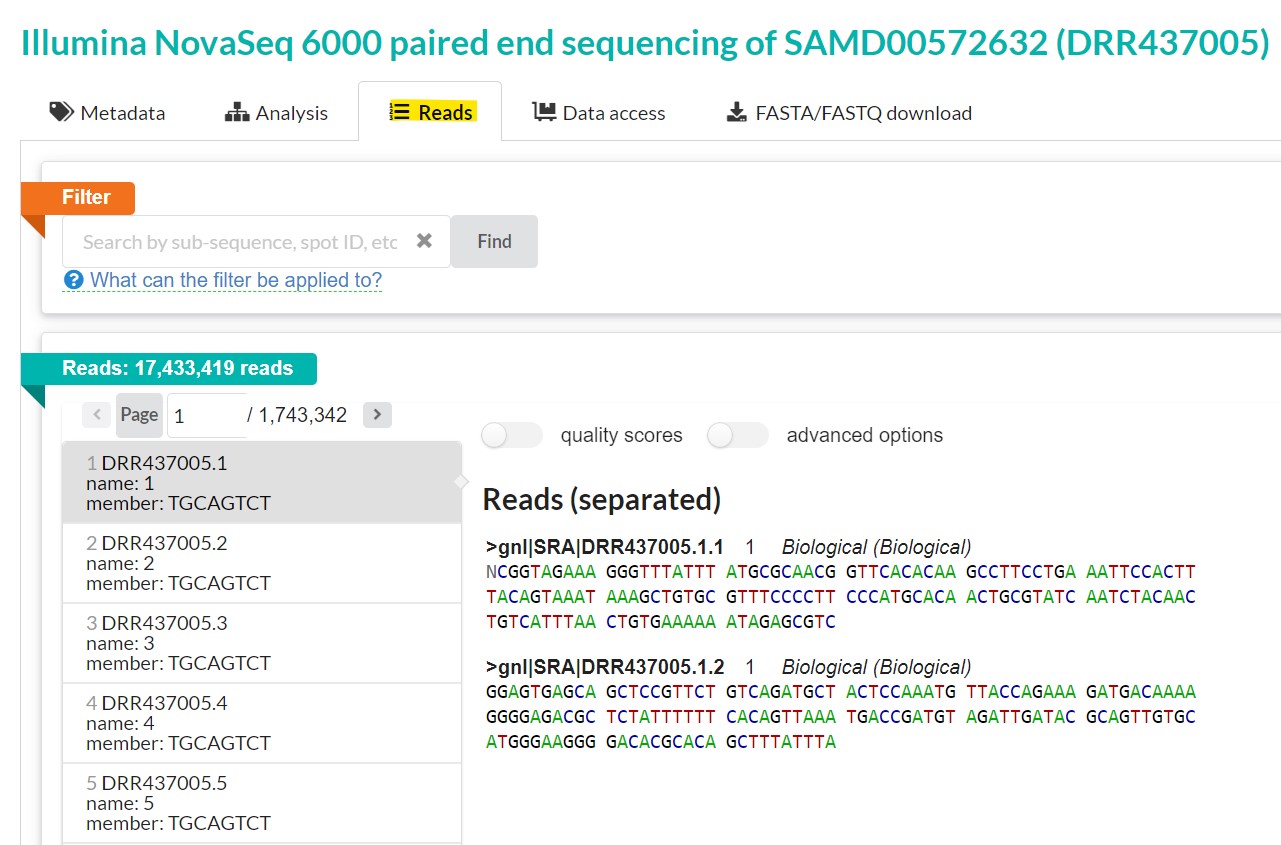
| You first need to get the individual read’s SRA number. To do this, go back to the “reads” tab. This file has 17,433,419 reads. The reads are listed on the left pane. Clicking on each read name gives you the paired end reads on your right. You can opt to turn on quality scores and to separate or join reads using the “advanced options” button. To download individual reads, get the read name and use the prefetch tool to download it. For instance, the read name for the first read is DRR437005.1 |
|
| Download the reads using the prefetch tool on your command line:
prefetch DRR437005.1 |
| Convert the SRA file to fastq using the following command:
fastq-dump DRR437005.1 This will generate a file called DRR437005.1.fastq |
| Congratulations! You have successfully retrieved sequences from the NCBI SRA and converted them into the .fastq format. We will now learn how to analyze such sequences.
Next, we look at how to create phylogenetic trees using Mr. Bayes, Fig Tree, and retrieved NCBI sequences. |

NEXT >> How to Create Phylogenetic Trees using Mr. Bayes and Fig Tree.